Spatial transcriptomics deconvolution visualization¶
We support to visualize the scatterpie chart for any deconvolution or label transferring tools.
Here we will describe how to use SPOTlight and Seurat Label transferring to visualize in stLearn
SPOTlight¶
You can follow the tutorial of SPOTlight in their git repository: https://github.com/MarcElosua/SPOTlight
After done the tutorial, you can run this R code to get deconvolution_result.csv
[1]:
# This is R code. You should run this after done SPOTlight tutorial
tmp = as.data.frame(decon_mtrx)
row.names(tmp) <- row.names(brain@meta.data)
write.csv(t(tmp[1:(length(tmp)-1)]),"deconvolution_result.csv")
Note that: - brain is the Seurat object of Spatial Transcriptomics - We save the decon_mtrx to .csv file as the input of our deconvolution visualization function
Seurat label transferring¶
Seurat provided a vignette about spatial transcriptomics analysis (https://satijalab.org/seurat/v3.2/spatial_vignette.html).
In the section: Integration with single-cell data, you can follow this to do the label transferring.
After that, you can run this code in R to get the deconvolution_result.csv
[1]:
# This is R code. You should run this after done Integration with single-cell data tutorial
write.csv(predictions.assay@data[-nrow(predictions.assay@data),],"deconvolution_result.csv")
Other software¶
If you use other software, you should convert the result to this format:
[1]:
deconvolution_result
[1]:
| AAACAAGTATCTCCCA-1 | AAACACCAATAACTGC-1 | AAACAGCTTTCAGAAG-1 | AAACAGGGTCTATATT-1 | AAACAGTGTTCCTGGG-1 | AAACATTTCCCGGATT-1 | AAACCGGGTAGGTACC-1 | AAACCGTTCGTCCAGG-1 | AAACCTAAGCAGCCGG-1 | AAACCTCATGAAGTTG-1 | ... | TTGTGTATGCCACCAA-1 | TTGTGTTTCCCGAAAG-1 | TTGTTAGCAAATTCGA-1 | TTGTTCAGTGTGCTAC-1 | TTGTTGGCAATGACTG-1 | TTGTTGTGTGTCAAGA-1 | TTGTTTCACATCCAGG-1 | TTGTTTCATTAGTCTA-1 | TTGTTTCCATACAACT-1 | TTGTTTGTATTACACG-1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B.cells | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.198548 | ... | 0.420818 | 0.486748 | 0.281131 | 0.000000 | 0.000000 | 0.000000 | 0.498129 | 0.000000 | 0.000000 | 0.626930 |
| BAM | 0.000000 | 0.000000 | 0.194210 | 0.000000 | 0.349818 | 0.174129 | 0.128605 | 0.124109 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.193690 | 0.272291 | 0.000000 | 0.000000 | 0.000000 | 0.177772 |
| cDC1 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.149757 | 0.000000 | 0.000000 | 0.000000 |
| cDC2 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.287142 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.186463 | 0.000000 | 0.000000 | 0.000000 |
| ILC | 0.247162 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.147220 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.192057 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| Microglia | 0.220380 | 0.166595 | 0.000000 | 0.181610 | 0.000000 | 0.000000 | 0.198072 | 0.000000 | 0.166483 | 0.172001 | ... | 0.320737 | 0.314146 | 0.000000 | 0.187380 | 0.234365 | 0.000000 | 0.165652 | 0.164594 | 0.229121 | 0.000000 |
| migDC | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.126951 | 0.000000 | 0.000000 | ... | 0.000000 | 0.199107 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| Monocytes.Mdc | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| Neutrophils | 0.207182 | 0.000000 | 0.182595 | 0.151981 | 0.000000 | 0.000000 | 0.000000 | 0.149797 | 0.349811 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.192915 | 0.195298 |
| NK.cells | 0.000000 | 0.000000 | 0.177223 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.219932 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| Others | 0.325277 | 0.441767 | 0.270445 | 0.344442 | 0.000000 | 0.484492 | 0.222825 | 0.324590 | 0.000000 | 0.410401 | ... | 0.258446 | 0.000000 | 0.253745 | 0.403855 | 0.193312 | 0.344915 | 0.000000 | 0.319064 | 0.267189 | 0.000000 |
| pDC | 0.000000 | 0.000000 | 0.000000 | 0.152278 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.293502 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.224080 | 0.000000 | 0.000000 | 0.000000 | 0.187533 | 0.143427 | 0.000000 |
| T.NKT.cells | 0.000000 | 0.212961 | 0.000000 | 0.169689 | 0.157046 | 0.167671 | 0.160222 | 0.146710 | 0.190204 | 0.000000 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.382794 | 0.000000 | 0.328808 | 0.000000 | 0.000000 |
| yd.T.cells | 0.000000 | 0.178677 | 0.175527 | 0.000000 | 0.205995 | 0.173708 | 0.143056 | 0.127842 | 0.000000 | 0.219050 | ... | 0.000000 | 0.000000 | 0.273067 | 0.184685 | 0.158700 | 0.000000 | 0.000000 | 0.000000 | 0.167349 | 0.000000 |
14 rows × 2968 columns
Note: - Columns are spot barcode - Rows are labels - Values are the proportion or probability value
stLearn deconvolution visualization¶
Running the basic analysis step of stLearn
[1]:
import stlearn as st
[2]:
data = st.Read10X(path="BRAIN_PATH")
data.var_names_make_unique()
st.pp.filter_genes(data,min_cells=3)
st.pp.normalize_total(data)
st.pp.log1p(data)
st.pp.scale(data)
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Normalization step is finished in adata.X
Log transformation step is finished in adata.X
Scale step is finished in adata.X
[3]:
st.em.run_pca(data,n_comps=10,random_state=0)
st.pp.neighbors(data,n_neighbors=15,use_rep='X_pca',random_state=0)
st.tl.clustering.louvain(data,random_state=0)
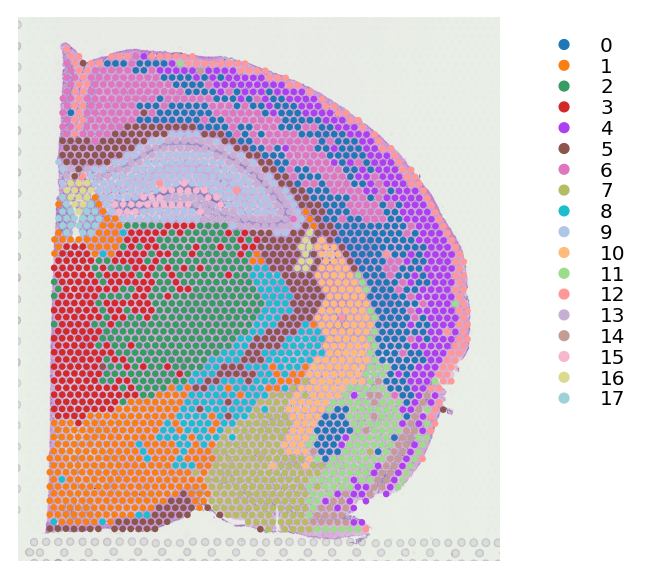
st.pl.cluster_plot(data,use_label="louvain",image_alpha=1,size=7)
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
Created k-Nearest-Neighbor graph in adata.uns['neighbors']
Applying Louvain clustering ...
Louvain clustering is done! The labels are stored in adata.obs['louvain']
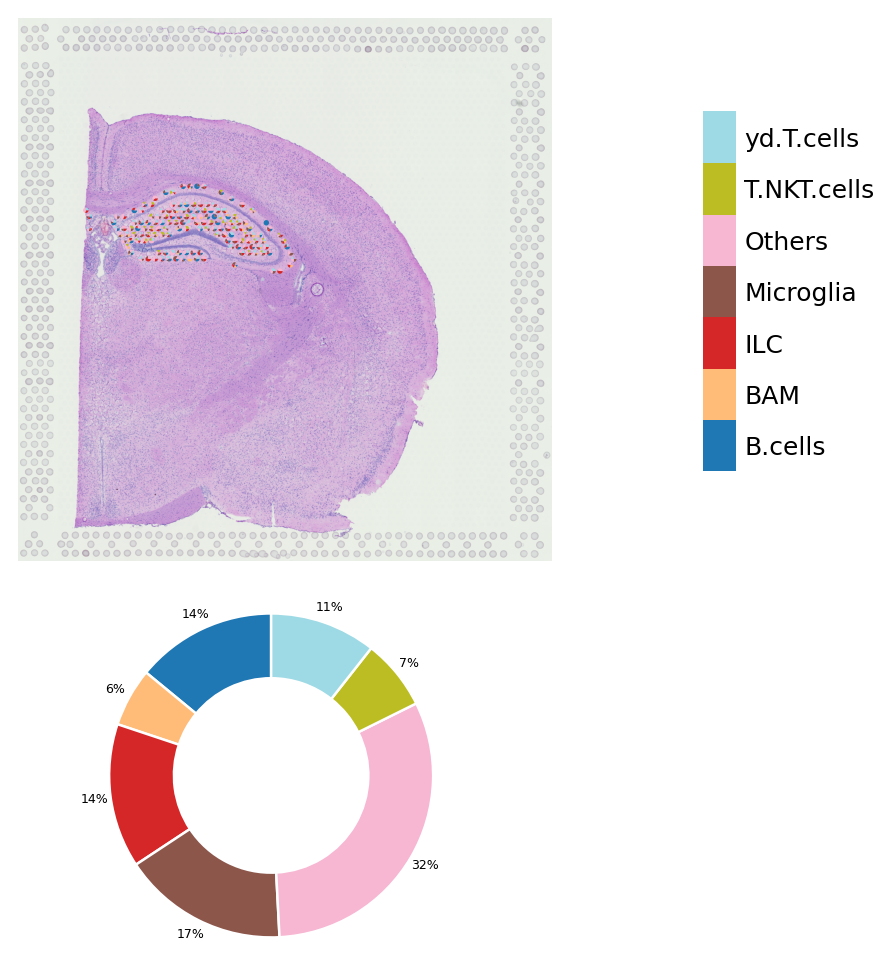
Add annotation from other software and visualize it into a scatter pie plot with donut chart
[4]:
st.add.add_deconvolution(data,annotation_path="deconvolution_result.csv")
The annotation is added to adata.obs['louvain_anno']
We also provide a threshold parameter that do the filtering based on quantile. The objective is removing the noise labels.
[5]:
st.pl.deconvolution_plot(data,threshold=0.5)
[6]:
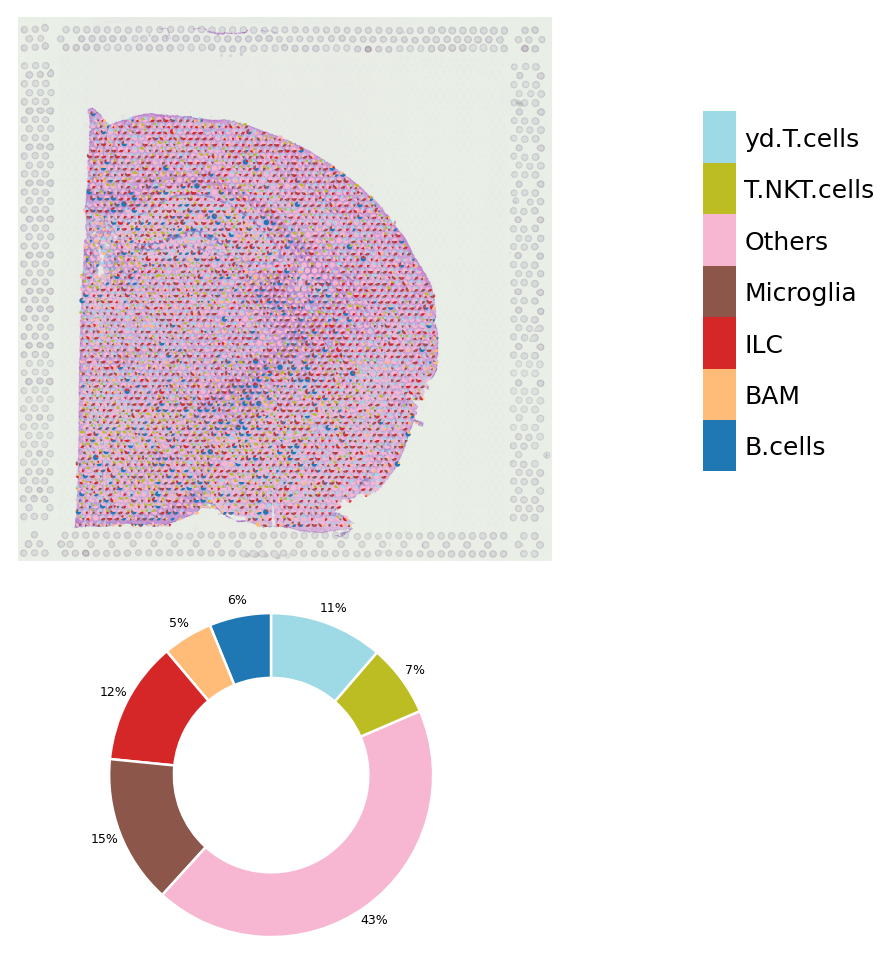
st.pl.deconvolution_plot(data,threshold=0.0)
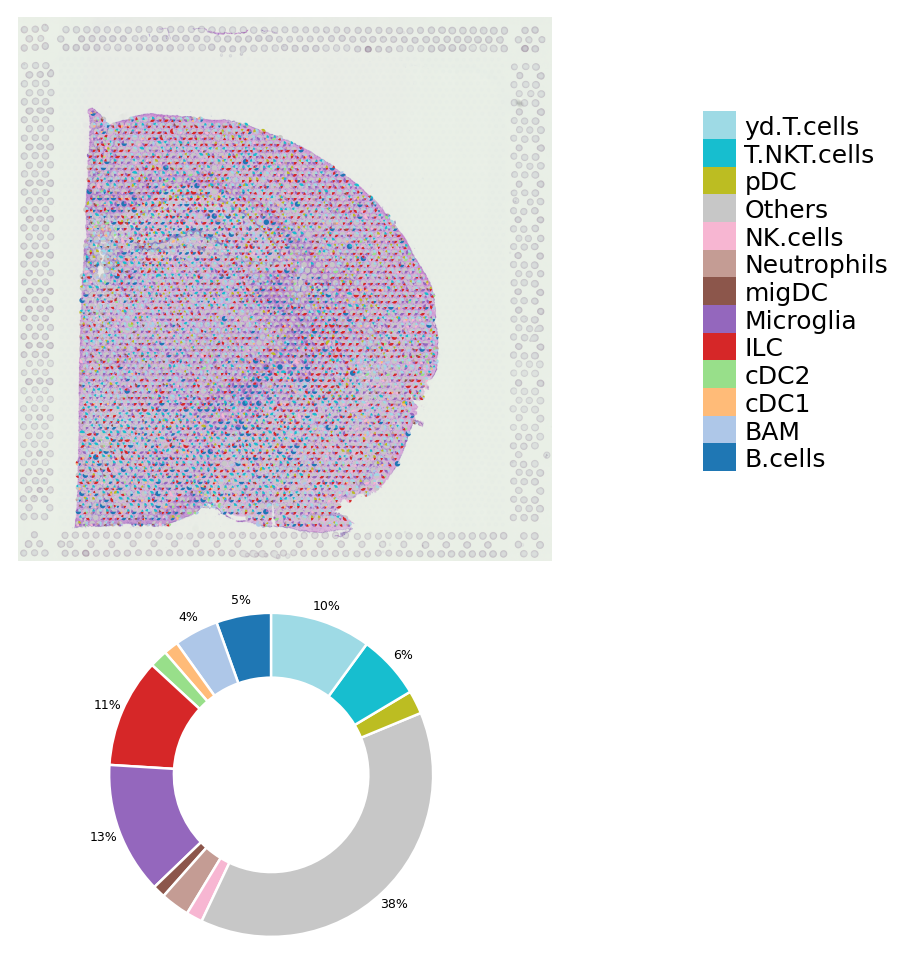
You also can examine the proportion of cell types in each cluster
[7]:
deconvolution_plot(data,cluster=9)