stSME Comparison¶
This tutorial shows the stSME normalization effect between of two scenarios:
Normal (without stSME)
stSME applied on raw gene counts
In this tutorial we use Mouse Brain (Coronal) Visium dataset from 10x genomics website.
[1]:
import os
import platform
# Only constrain threads on macOS where BLAS/numba deadlocks are common.
# Must run before any numpy/scanpy import.
if platform.system() == "Darwin":
os.environ["OPENBLAS_NUM_THREADS"] = "1"
os.environ["MKL_NUM_THREADS"] = "1"
os.environ["NUMBA_NUM_THREADS"] = "1"
n_cpus = 1
else:
n_cpus = None
[2]:
import scanpy as sc
import stlearn as st
import pathlib as pathlib
import numpy as np
import random as random
import os as os
st.settings.set_figure_params(dpi=120)
# Make sure all the seeds are set
seed = 0
np.random.seed(seed)
random.seed(seed)
os.environ['PYTHONHASHSEED'] = str(seed)
# Ignore all warnings
import warnings
warnings.filterwarnings("ignore")
[3]:
st.settings.datasetdir = pathlib.Path.cwd().parent / "data"
[4]:
data = sc.datasets.visium_sge(sample_id="V1_Adult_Mouse_Brain")
data = st.convert_scanpy(data)
[5]:
# pre-processing for gene count table
st.pp.filter_genes(data, min_cells=1)
st.pp.normalize_total(data)
st.pp.log1p(data)
Normalization step is finished in adata.X
Log transformation step is finished in adata.X
[6]:
# pre-processing for spot image
st.pp.tiling(data, out_path="tiling")
# this step uses deep learning model to extract high-level features from tile images
# may need few minutes to be completed
st.pp.extract_feature(data)
Tiling image: 100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████ [ time left: 00:00 ]
Extract feature: 100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████▉ [ time left: 00:00 ]
The morphology feature is added to adata.obsm['X_morphology']!
[7]:
# run PCA for gene expression data
st.em.run_pca(data, n_comps=50)
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
Normal¶
[8]:
data_normal = data.copy()
stSME¶
[9]:
data_sme = data.copy()
# apply stSME to normalise log transformed data
st.spatial.sme.sme_normalize(data_sme, use_data="raw")
data_sme.X = data_sme.obsm['raw_SME_normalized']
st.em.run_pca(data_sme, n_comps=50)
Adjusting data: 100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████ [ time left: 00:00 ]
The data adjusted by SME is added to adata.obsm['raw_SME_normalized']
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
[10]:
import matplotlib.pyplot as plt
def plot_comparison(i):
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
# Plot into the axes but suppress individual titles
st.pl.gene_plot(data_normal, gene_symbols=i, size=3, ax=axes[0])
st.pl.gene_plot(data_sme, gene_symbols=i, size=3, ax=axes[1])
# Remove individual titles set by the function
axes[0].set_title("")
axes[1].set_title("")
# Set subplot labels and shared title
axes[0].set_title("Normal", fontsize=12)
axes[1].set_title("stSME", fontsize=12)
fig.suptitle(i, fontsize=16, fontweight="bold")
plt.tight_layout()
plt.show()
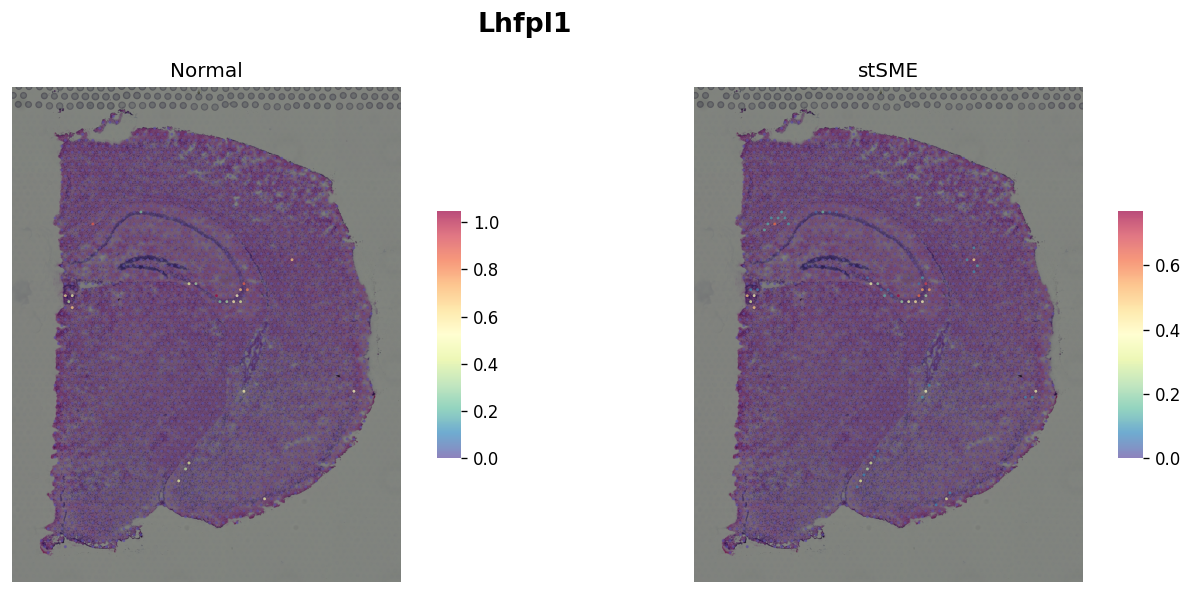
Marker gene for CA3¶
[11]:
plot_comparison("Lhfpl1")
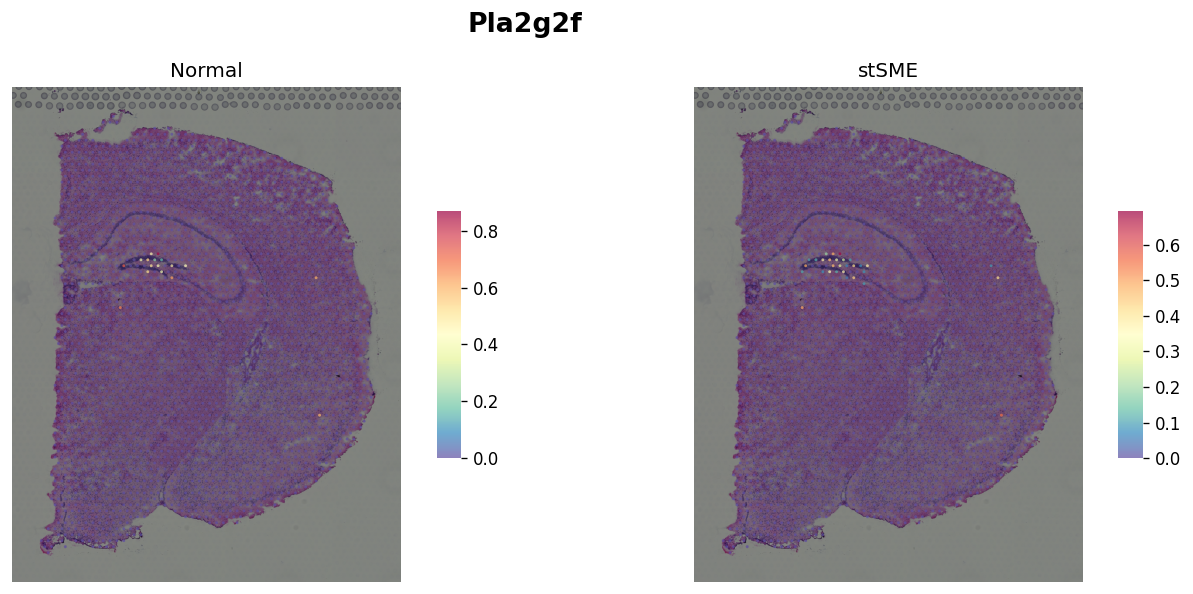
Marker gene for DG¶
[12]:
plot_comparison("Pla2g2f")
Tutorial by Xiao Tan